
More Information
Submitted: March 29, 2024 | Approved: April 11, 2024 | Published: April 12, 2024
How to cite this article: López-Chávez E, García-Quiroz A, Peña-Castañeda YA, Díaz-Góngora JAI, Mendoza-Espinoza JA, et al. Sites and Zones of Maximum Reactivity of the most Stable Structure of the Receptor-binding Domain of Wild-type SARS-CoV-2 Spike Protein: A Quantum Density Functional Theory Study. J Clin Intensive Care Med. 2024; 9: 008-016.
DOI: 10.29328/journal.jcicm.1001047
Copyright license: © 2024 López-Chávez E, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Highest occupied molecular orbital; Lowest unoccupied molecular orbital; Density functional theory; Receptor binding domain; Severe acute respiratory syndrome coronavirus 2; Corona virus disease of 2019

Sites and Zones of Maximum Reactivity of the most Stable Structure of the Receptor-binding Domain of Wild-type SARS-CoV-2 Spike Protein: A Quantum Density Functional Theory Study
Ernesto López-Chávez1* , Alberto García-Quiroz1, Yesica Antonia Peña-Castañeda1, José Antonio Irán Díaz-Góngora2, José Alberto Mendoza-Espinoza1, Jose Antonio López-Barrera1 and Fray de Landa Castillo-Alvarado3
, Alberto García-Quiroz1, Yesica Antonia Peña-Castañeda1, José Antonio Irán Díaz-Góngora2, José Alberto Mendoza-Espinoza1, Jose Antonio López-Barrera1 and Fray de Landa Castillo-Alvarado3
1Universidad Autónoma de la Ciudad de México, Fray Servando Teresa de Mier #92, Col. Obrera, Alc. Cuauhtémoc, C.P. 06080, Ciudad de México
2Centro de Investigación en Ciencia Aplicada y Tecnología Avanzada, Instituto Politécnico Nacional, Unidad Legaria, Calz. Legaria #694, Col. Irrigación, Alc. Miguel Hidalgo, C.P. 11500, Ciudad de México
3Escuela Superior de Física y Matemáticas, Instituto Politécnico Nacional, Edificio 9, Col. Lindavista, Alc. Gustavo A. Madero, C.P 07738, Ciudad de México
*Address for Correspondence: Ernesto López-Chávez, Universidad Autónoma de la Ciudad de México, Fray Servando Teresa de Mier #92, Col. Obrera, Alc. Cuauhtémoc, C.P. 06080, Ciudad de México, Email: [email protected]; [email protected]
Today, it is well known that Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has four types of proteins within its structure, between them the spike protein (S). The infection mechanism is carried out by the entry of the virus into the human host cell through the S protein, which strongly interacts with the human cell receptor angiotensin-converting enzyme 2 (ACE2). In this work, we propose an atomic model of the Receptor Binding Domain (RBD) of the S spike protein of the wild-type SARS-CoV-2 virus. The molecular structure of the model was composed of 50 amino acids that were chemically bonded, starting with Leucine and ending with one amino acid Tyrosine. The novelty of our work lies in the importance of knowing the sites and zones of maximum reactivity of the RBD from the fundamental levels of quantum mechanics considering the atomic structure of matter. For this, the local and global reactivity indices of the RBD were calculated, such as frontier orbitals, Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), Fukui indices, chemical potential, chemical hardness, electrophilicity index; with this, it will be possible to know what type of molecules are more likely to interact with the RBD structure, and in this way, new knowledge will be generated at the quantum, atomic and molecular level to inhibit the virulent effects of wild-type SARS-CoV-2. Finally, in order to identify the functional groups within the most stable structure and thereby verify the future reactions that can be carried out between the RBD structure and biomolecules, the Infrared (IR) absorption spectrum was calculated. For this work, we used Material Studio v6.0 which uses the density functional theory (DFT) implemented in its DMol3 computational code. The IR spectrum was obtained using the Spartan ‘94 computer code. One novelty would be that we found nine amino acids more that could make the RBD and ACE2 binding further the already known. Thus, the Mulliken charge distribution indicates that the highest concentrations of positive and negative charge are found in the zones 477S, 478T, 484E, and 501N amino acids letting ionic or Van der Waals possible interactions with other structures.
Undoubtedly, there have been notable advances in combating the coronavirus disease of 2019 (COVID-19) pandemic, which has affected the health of a good percentage of the world’s inhabitants and unfortunately caused the death of many of them, however, until June 14, 2022, around 536.6 million cases of SARS-CoV-2 have been registered in the world. The coronavirus that originated in the Chinese city of Wuhan has spread to all countries in the geography of Latin America and the world [1,2]. So, there is still much to know about the development, changes, and mutations, as well as the effects on the human body of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) virus and its different mutations, which are causes of the symptoms of the COVID-19.
Nowadays, it is well known that SARS-CoV-2 has four types of proteins within its structure, namely the spike protein (S), the membrane protein (M), the envelope protein (E), and the nucleocapsid protein (N). The mechanism of infection is carried out by the entry of the virus into the human host cell through protein S, which strongly interacts with the human cell receptor angiotensin-converting enzyme 2 [3,4]. The structure of the S protein is made up of more than 1200 amino acids divided into two functional subunits called S1 and S2. Within S1 is the receptor binding domain (RBD) consisting of 50 amino acids from L455 (Leucine) to Y505 (Tyrosine). It is precisely the RBD that is directly responsible for binding strongly to ACE2. For this reason, many of the interdisciplinary research groups around the world have focused their efforts on studying the structure and properties of RBD together with the mechanisms of interaction with ACE2 [5-9].
Due to the experimental difficulty in determining structural properties at quantum levels, some studies have carried out research using modeling techniques and computational simulations to determine, among other things, the binding mechanism of SARS-CoV-2 to ACE2, which may aid the design of therapeutics for COVID-19 infection [10,11]. However, the models used for the RBD in these studies do not consider the atomic structure of the amino acids nor do they locate the points and zones of maximum reactivity within the RBD. That is why more research effort is required to answer several questions about the mechanisms of infection by SARS-CoV-2 related to the reactivity and selectivity indices of the RBD of the protein of the spike S of SARS-CoV-2 [8,12-15].
In this work, in order to find the points and zones of maximum reactivity of the RBD, an atomic model of the RBD of SARS-CoV-2 was built by chemically joining the 50 amino acids that form it and the most stable structure was obtained by carrying out the process energy minimization. It is well known that the full length of S has more than 1200 amino acids, which is divided into two subunits (S1 and S2), within subunit 1 the Receptor binding domain from L455 to Y505 is located. In this work, only this region was studied since it is the most prone to bind with the human cell receptor angiotensin-converting enzyme 2. The results of energy minimization and normal vibration modes (all being of positive frequency) guarantee that the atomic model is stable and is a good approximation to the real structure of SARS-CoV-2 S spike-RBD. Subsequently, using density functional theory techniques, the HOMO-LUMO frontier orbitals were calculated along with their respective energies, as well as the Fukui indices and the charge distribution through the Mulliken population analysis.
The results obtained with this alternative model, which is based on quantum, atomic, and molecular levels, coincide with what has been reported in the scientific literature since 2020; moreover, we gain new insight into new sites and zones in RBD with a high probability of binding to other molecules. The new knowledge generated at this may be used in different endeavors to inhibit the virulent effects of wild-type SARS-CoV-2 and new variants.
The first step of this work consisted of building the atomic model of the RBD region of the Sars-CoV-2 S-protein, for which the construction tools of the Materials Studio software were used; see Figure A1 and Table A2 of the appendix for easier reproducibility. The atomic structure of the RBD consists of a set of 50 chemically linked amino acids according to the sequence L (Leucine 455), F, R, K, S, N, L, K, P, F, E, R, D, I, S, T, E, I, Y, Q, A, G, S, T, P, C, N, G, V, E, G, F, N, C, Y, F, P, L, Q, S, Y, G, F, Q, P, T, N, G, V, G, Y (Tyrosine 505), each letter corresponds to a type of amino acid [16]. Subsequently, optimization of the geometry and minimization of the energy of the entire RBD was carried out in order to obtain the most stable structure.
The calculation of HOMO and LUMO molecular orbitals, as well as the Fukui indices and the Mulliken charge population, were performed within the Density Functional Theory implemented in the DMol3 computational code from Materials Studio. Quantum exchange-correlation interactions were calculated using the generalized gradient approximation (GGA) and the Perdew-Wang 91 (PW91) functional [17-19]. Numerical functions of the Double Numerical plus d-Functions (DND) type with unrestricted spin were used as the base set. Pseudopotentials were not used for these calculations [20]. The entire RBD system was studied electrically neutral and within an aqueous medium of the dielectric constant of 78.54 belonging to water. The convergence parameters of the variables in the geometry optimization process were established as follows: energy, 10-4 Ha; gradient, 2x10-2 Å; displacement, 5 x 10-2 Å; maximum displacement, 0.3 Å.
With that information, the electron affinity (EA) and ionization potential (IP) were obtained through electronic energies of the anionic, neutral, and cationic states of the RBD, symbolized as E(N+1), E(N), and E(N-1), respectively. The equations for electron affinity and ionization potential are given by:
(1)
and
(2)
According to Koopmans theorem, those energies can be used to calculate global reactivity indices such as chemical potential (µ), electrophilicity index (ꞷ), and chemical hardness (η). To calculate these global properties of reactivity, the finite difference approximation must be used since the derivative of the energy with respect to N is discontinuous, in this way, we obtain the following formulas [21-22]:
(3)
(4)
(5)
The reactivity and selectivity of the molecular structure of the RBD are fully characterized through local and global electronic properties. To find the local selectivity or reactivity of the RBD molecule, the Fukui functions were used. These functions were introduced by Parr and Yang [24] as a generalization of the frontier orbital reactivity concepts, and they represent the sensitivity of the chemical potential due to an external disturbance, as well as the change in electron density when the number of electrons changes. So, by definition, the Fukui functions are expressed as [25]:
(6)
Applying the finite difference approximation and the Mulliken scheme for a system containing N electrons, the numerical value of the Fukui functions at all k-sites of our molecule for attack by a nucleophile, an electrophile, and a radical are obtained, respectively, with the formulas [26]:
7)
8)
(9)
Where qk(N+1), qk(N), qk(N-1) are the charge in the k-site when the system there are N+1, N, N-1 electrons.
Since the Fukui indices represent the variation of the local electron density induced by the introduction of electrons into the system or the escape of electrons from the system, the greater this variation, the more reactive the system will be at position r of the molecule, since A chemical potential gradient will be generated that will cause a greater charge transfer. So, the higher the numerical value of the f(r), the greater the reactivity we will have on the site.
The latter will provide more elements to predict future mutations or drug design to inhibit SARS-CoV-2.
Finally, in order to obtain the absorption spectrum of the RBD, the structure was optimized using the Molecular Mechanics Merck Molecular Force Field (MMFF) level of theory, using the Spartan ‘94 program.
Figure 1, and Figure A1 of the appendix, correspond to the most stable structure of the RBD quantum model. It is made up of 786 atoms, of which 380 are hydrogen (H, white), 260 are carbon (C, gray), 78 are oxygen (O, red), 66 are nitrogen (N, green), and 2 are sulfur (S, yellow). The sequence of amino acids, together with any properties of them, which form the RBD system, is presented below:
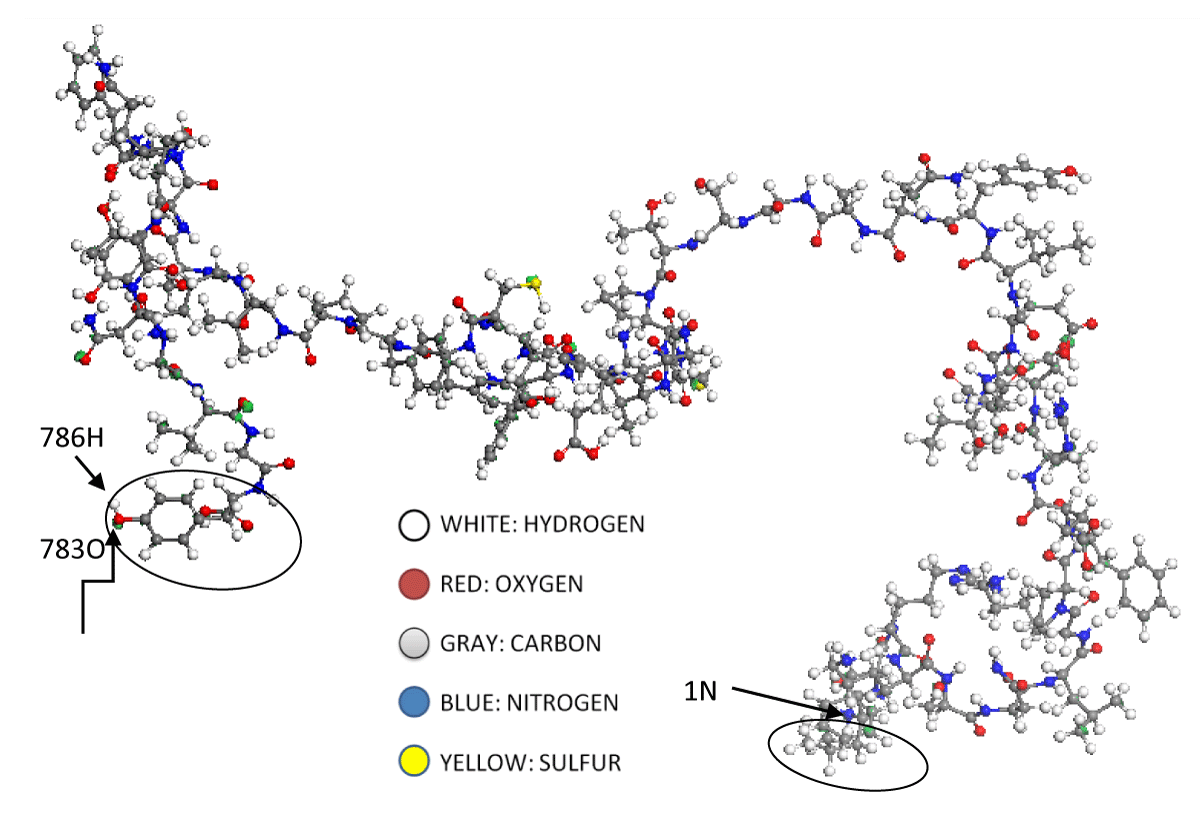
Figure 1: At the RBD molecular model, left side, the 783 oxygen, and 786 hydrogen atoms respectively are observed, see attached table A1. These two atoms belong to the tyrosine amino acid, cited in attached table A2. This amino acid is circled on the left side of Figure 1. Moreover, we can say that the last amino acid, circled on the right side of this figure is the Leucine amino acid. Beginning with a blue nitrogen atom, numbered as 1N atom; go to the attached table A2 and figure A1.
455 L, Leucine (hydrophobic-nonpolar-nullification of forces, Hydrophobic interactions, and Van der Waals)
456 F, Phenylalanine (aromatic-hydrophobic-nonpolar-most reactive, Van Der Waals interactions with receptor. Hydrophobic and Van der Waals interactions)
457 R, Arginine (hydrophilic-unstable)
458 K, Lysine (hydrophilic-unstable)
459 S, Serine (hydrophilic-polar-reactive-unstable in alkaline medium)
460 N, Asparagine (hydrophilic-negative charge-can be ionized or non-charge responsible)
461 L, Leucine (hydrophobic-nonpolar-nullification of forces)
462 K, Lysine (hydrophilic-unstable)
463 P, Proline (nonpolar-but not hydrophobic)
464 F, Phenylalanine (aromatic-hydrophobic-nonpolar-most reactive)
465 E, Glutamic acid (hydrophilic-negative charge- can be ionized or not-responsible for charge)
466 R, Arginine (hydrophilic-unstable)
467 D, Aspartic acid (hydrophilic-negative charge- can be ionized or not-responsible for charge)
468 I, Isoleucine (hydrophobic)
469 S, Serine (hydrophilic-polar-reactive-unstable in alkaline medium)
470 T, Threonine (hydrophilic-polar-reactive-unstable in alkaline medium)
471 E, Glutamic acid (hydrophilic-negative charge- may be ionized or non-charge responsible)
472 I, Isoleucine (hydrophobic)
473 Y, Tyrosine (aromatic-hydrophobic-nonpolar-most reactive) Van Der Waals interactions with receptor
474 Q, Glutamine (hydrophilic-negative charge-can be ionized or non-charge responsible)
475 A, Alanine (hydrophobic)
476 G, Glycine (hydrophobic) Reported virus/receptor recognition (weaker)
477 S, Serine (hydrophilic-polar-reactive-unstable in alkaline medium). Mutations: N, K
478 T, Threonine (hydrophilic-polar-reactive-unstable in alkaline medium) Mutations: K
479 P, Proline (nonpolar-but not hydrophobic)
480 C, Cysteine (unstable under S-oxidation conditions)
481 N, Asparagine (hydrophilic-negative charge- may be ionized or non-charge responsible)
482 G, Glycine (hydrophobic)
483 V, Valine (hydrophobic)
484 E, Glutamic acid (hydrophilic-negative charge-can be ionized or non-charge responsible) Mutations: K, K, K
485 G, Glycine (hydrophobic)
486 F, Phenylalanine (aromatic-hydrophobic-nonpolar-most reactive)
487 N, Asparagine (hydrophilic-negative charge-may ionize or not-responsible for charge) Virus/receptor recognition reported
488 C, Cysteine (unstable under S-oxidation conditions)
489 Y, Tyrosine (aromatic-hydrophobic-nonpolar-most reactive) Reported virus/receptor recognition (weak)
490 F, Phenylalanine (aromatic-hydrophobic-nonpolar-most reactive)
491 P, Proline (nonpolar-but not hydrophobic)
492 L, Leucine (hydrophobic-nonpolar-nullification of forces)
493 Q, Glutamine (hydrophilic-negative charge-can be ionized or non-charge responsible)
494 S, Serine (hydrophilic-polar-reactive-unstable in alkaline medium)
495 Y, Tyrosine (aromatic-hydrophobic-nonpolar-most reactive)
496 G, Glycine (hydrophobic)
497 F, Phenylalanine (aromatic-hydrophobic-nonpolar-most reactive)
498 Q, Glutamine (hydrophilic-negative charge-may be ionized or non-charge responsible) Virus/receptor recognition reported
499 P, Proline (nonpolar-but not hydrophobic)
500 T, Threonine (hydrophilic-polar-reactive-unstable in alkaline medium) Reported virus/receptor recognition
501 N, Asparagine (hydrophilic-negative charge- can be ionized or not-responsible for charge). Mutations: Y, Y, Y, Y.
502 G, Glycine (hydrophobic) Virus/receptor recognition reported.
503 V, Valine (hydrophobic)
504 G, Glycine (hydrophobic)
505 Y, Tyrosine (aromatic-hydrophobic-nonpolar-most reactive)
The type of atom, the assigned number, and their respective coordinates are reported in Table A1 of the appendix. The sequence of amino acids together with the atoms (identified with their assigned number in Table A1) that constitute each one of them is summarized in Table A2) of the appendix.
| Table 1: Sites with the highest probability of electrophilic attacks (f-) within the HOMO frontier molecular orbital, together with the sites with the highest probability of nucleophilic attacks (f+) located within the LUMO frontier orbital, and the sites with the highest probability of attacks with radicals (f0) along the RBD molecule. The Mulliken charges and the position coordinates of the respective atoms are also reported. | ||||
| Frontier molecular orbital | Indexed Atom | Fukui index | Mulliken atomic charge (a.u) | Coordinates (x(A),y (A),z (A)) |
| HOMO | O(528) | f-= 0.008 | -0.499 | (26.31, -34.20, 40.76) |
| HOMO | C(533) | f-= 0.007 | -0.245 | (24.40, -30.08, 45.43) |
| HOMO | C(534) | f-= 0.004 | -0.214 | (24.25, -30.66, 44.18) |
| HOMO | 0(542) | f-= 0.005 | -0.592 | (24.91, -30.32, 47.76) |
| LUMO | H(246) | f+= 0.023 | 0.227 | (55.71, -63.98, 65.92) |
| LUMO | C(257) | f+= 0.020 | 0.464 | (54.65, -60.42, 65.79) |
| LUMO | H(242) | f+= 0.012 | 0.188 | (55.43, -66.36, 65.28) |
| LUMO | C(238) | f+= 0.015 | 0.487 | (55.17, -63.21, 64.01) |
| NONE | 0(112) | f0= 0.004 | -0.525 | (54.57, -80.14, 75.02) |
| NONE | 0(684) | f0= 0.004 | -0.505 | (27.44, -26.71, 19.92) |
| NONE | S(417) | f0= 0.006 | -0.243 | (43.53, -54.73, 52.40) |
| NONE | 0(729) | f0= 0.004 | -0.508 | (15.71, -32.17, 25.64) |
On the other hand, the proper function of the HOMO frontier molecular orbital is shown in Figure 2. In this image, it can be seen that the HOMO orbital encloses the amino acid 489 Tyrosine; in addition, it is found that the eigenvalue of the HOMO is -5.559 eV, which indicates that the RBD can spontaneously donate electrons to other molecular structures whose LUMO has an energy lower than -5.559 eV. Thus, it is found that some of the regions with the greatest probability of reacting or interacting with other molecular structures, such as the ACE2 receptor cells of the human organism, is the amino acid 489 Tyrosine.
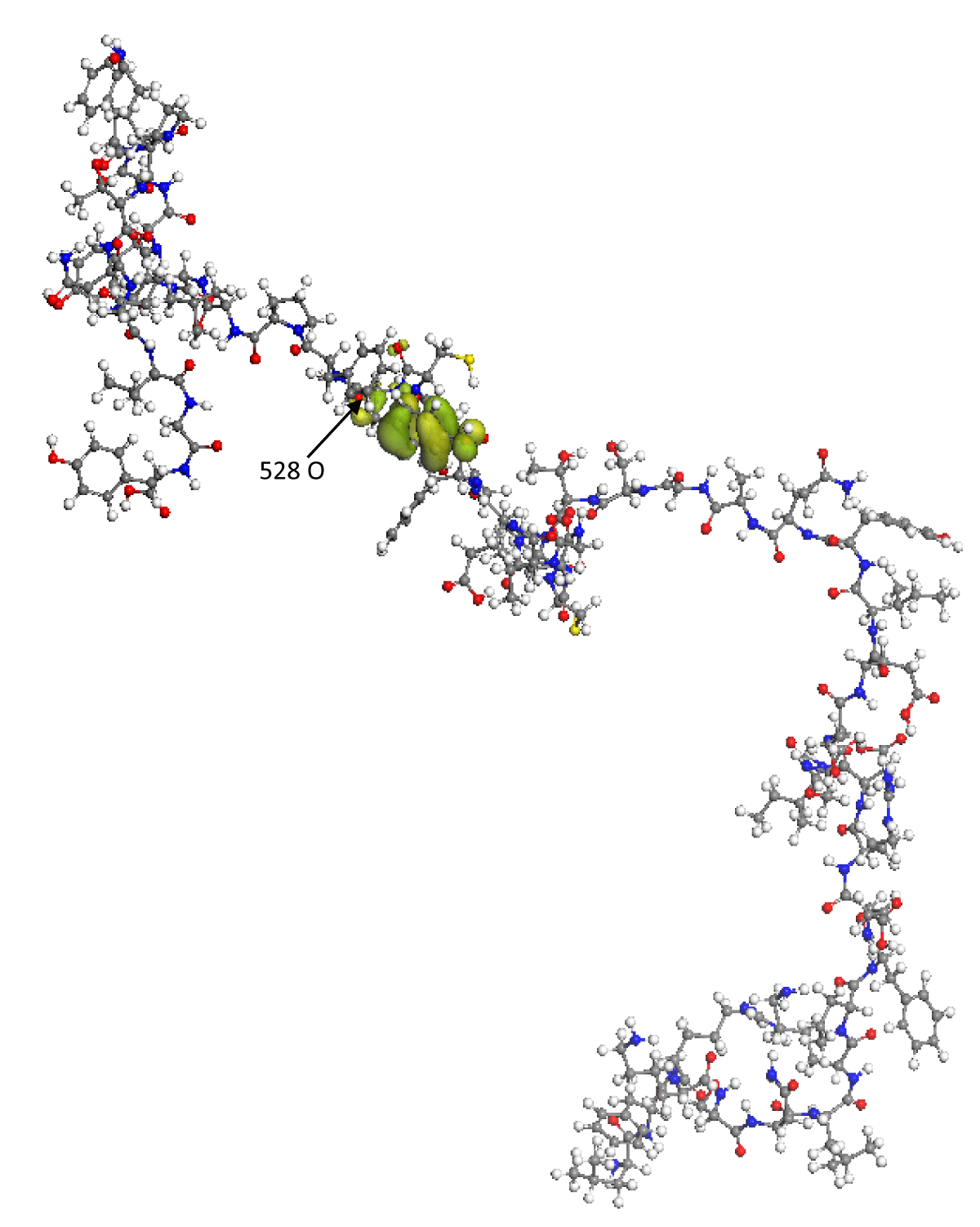
Figure 2: The green lobes represent the spatial distribution of the proper function of the HOMO frontier molecular orbital. The O(528) oxygen atom has the maximum Fukui index value.
Figure 3 represents the spatial distribution of the LUMO frontier molecular orbital, this orbital encloses the amino acids 467 Aspartic, 468 Isoleucine, 469 Serine, 470 Threonine, with its calculated eigenvalue being -2.167 eV, so that in these regions, the RBD molecule will be able to accept electrons from external molecular structures that have their HOMO energy greater than -2.167 eV. It is precisely in these areas determined by the HOMO and LUMO orbitals where there is a greater probability of presenting various mutations in the wild-type SARS-CoV-2 or failing that, react with other molecular structures that can inhibit them. The ELUMO-EHOMO energy gap is 3.392 eV, since the said gap is relatively high, it is found that the RBD nanostructure is highly internally stable and also behaves as an electrically non-conductive structure.
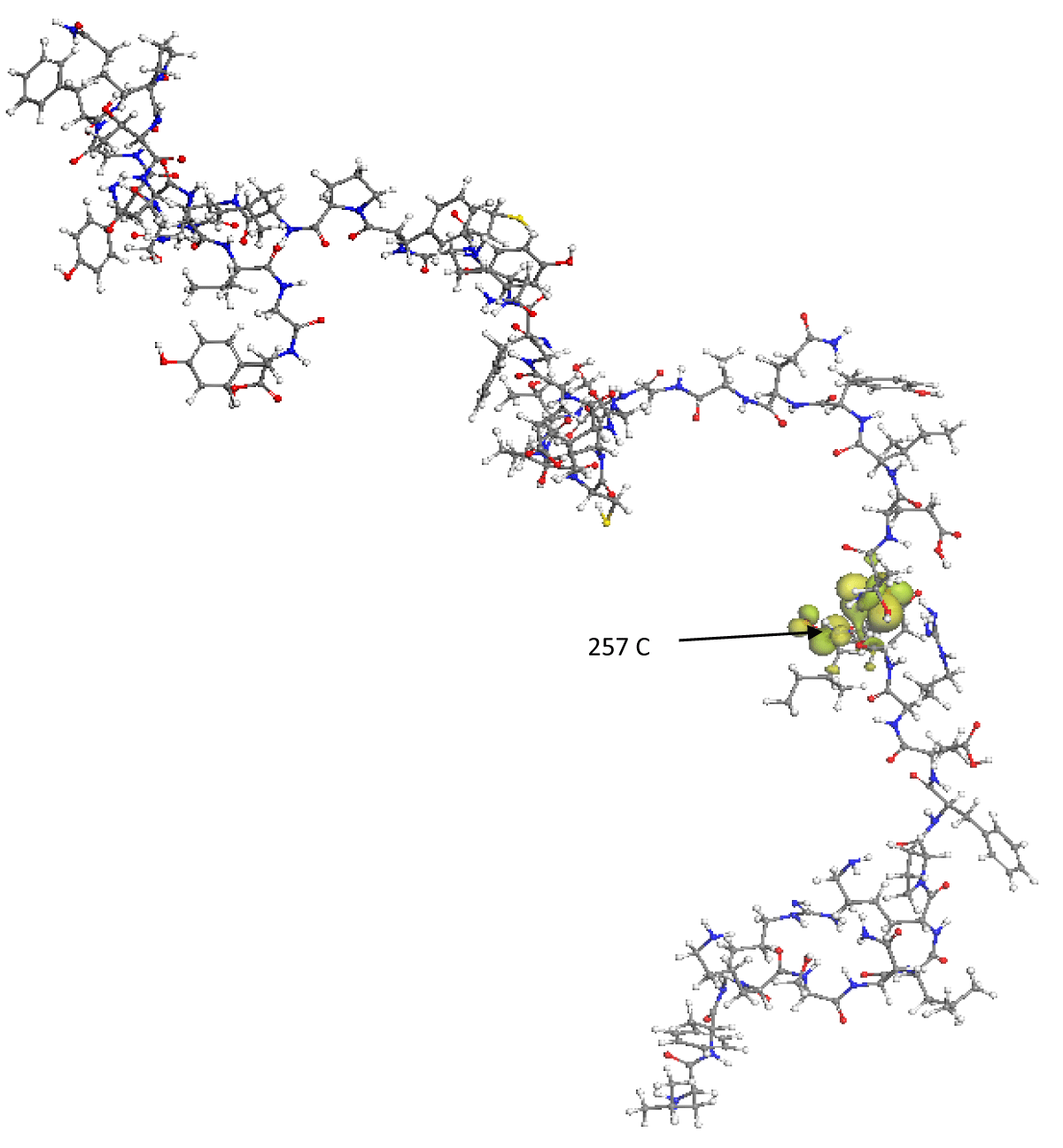
Figure 3: Spatial distribution of the LUMO frontier molecular orbital is represented by green lobes. The C(257) carbon atom has the maximum Fukui index value.
Using equations 1 to 5, we obtained the electronic energies values and the global reactivity indices given by E(N+1)= -555,745.03 eV, E(N)= -555, 748.30 eV, E(N-1)= -555,752.08 eV; EA= -3.27 eV, IP= 3.78 eV; µ= -0.255 eV, ω= 0.00922 eV, η= 3.525 eV.
The electronic chemical potential is a property of global chemical reactivity of the system which measures the tendency of electrons to escape from the system in equilibrium so that electrons flow from systems of high chemical potential to systems of low chemical potential, the flow will occur until the chemical potential of the two systems is equal. According to the previous results, it is found that the RBD will have a higher probability of reacting with other molecules or amino acids whose chemical potential differs markedly at -0.255 eV. This value of the chemical potential indicates that the RBD will have a high probability of transferring electrons or electric charge towards external molecules with a chemical potential lower than -0.255 eV, thus being able to behave as a Lewis base; while it will accept electrons from external molecules whose chemical potential is greater than -0.255 eV, being able to behave in this case as a Lewis acid. It is a condition for the RBD of the spike protein may mutate or react with other molecular structures that may belong to new drugs. In addition, the value of the chemical potential obtained in this work is an indicator of the high contagion or pandemic capacity of wild-type SARS-CoV-2.
On the other hand, equation 5 tells us that the chemical hardness is a global property of the system and measures the resistance that it imposes to the change in its electronic distribution. In this context, hardness is a descriptor of reactivity. According to the numerical value of the chemical hardness calculated in this work (3.525 eV) and its interpretation as the variation of the chemical potential with respect to the change in the number of electrons, we find that the molecular structure of the RBD remains almost invariant globally by carrying out chemical reactions with external systems or molecules. This could indicate that the global molecular structure of the RBD of the SARS-CoV-2 spike protein is resistant to being altered or modified, hence the relatively long time it took to have the first vaccine for the treatment of COVID-19.
The electrophilicity index is a measure of the energy stabilization of the system when it is saturated with electrons from the external medium. As the numerical value of the electrophilicity index of the RBD is relatively very small (0.0922 eV) we conclude that the RBD molecule is energetically very stable when chemical reactions with external molecules are carried out. This indicates that said molecule does not significantly alter its energy power during the reactions. This explains the high virulence rate of wild-type SARS-CoV-2, which is related to its pathogenicity.
The study of local selectivity or reactivity for the RBD molecule was done by Fukui functions using equations 6 to 9. We know that when the system undergoes a nucleophilic attack, the electrons enter the molecule in the LUMO orbital zone; while in electrophilic attacks, the electrons that are transferred come from the HOMO orbital zone. To relate what was said in the previous paragraph with the Fukui indices, in Table 1 we present the atoms of the LUMO and HOMO zones that have the largest value of their respective Fukui indices together with their respective charges, obtained by Mulliken’s population analysis. In addition, this table shows the atoms with the highest Fukui index for radical attacks along with their respective charge.
| Table 1: Sites with the highest probability of electrophilic attacks (f-) within the HOMO frontier molecular orbital, together with the sites with the highest probability of nucleophilic attacks (f+) located within the LUMO frontier orbital, and the sites with the highest probability of attacks with radicals (f0) along the RBD molecule. The Mulliken charges and the position coordinates of the respective atoms are also reported. | ||||
| Frontier molecular orbital | Indexed Atom | Fukui index | Mulliken atomic charge (a.u) | Coordinates (x(A),y (A),z (A)) |
| HOMO | O(528) | f-= 0.008 | -0.499 | (26.31, -34.20, 40.76) |
| HOMO | C(533) | f-= 0.007 | -0.245 | (24.40, -30.08, 45.43) |
| HOMO | C(534) | f-= 0.004 | -0.214 | (24.25, -30.66, 44.18) |
| HOMO | 0(542) | f-= 0.005 | -0.592 | (24.91, -30.32, 47.76) |
| LUMO | H(246) | f+= 0.023 | 0.227 | (55.71, -63.98, 65.92) |
| LUMO | C(257) | f+= 0.020 | 0.464 | (54.65, -60.42, 65.79) |
| LUMO | H(242) | f+= 0.012 | 0.188 | (55.43, -66.36, 65.28) |
| LUMO | C(238) | f+= 0.015 | 0.487 | (55.17, -63.21, 64.01) |
| NONE | 0(112) | f0= 0.004 | -0.525 | (54.57, -80.14, 75.02) |
| NONE | 0(684) | f0= 0.004 | -0.505 | (27.44, -26.71, 19.92) |
| NONE | S(417) | f0= 0.006 | -0.243 | (43.53, -54.73, 52.40) |
| NONE | 0(729) | f0= 0.004 | -0.508 | (15.71, -32.17, 25.64) |
From Table 1 we found that the sites most likely to suffer nucleophilic attacks into LUMO are those occupied by the H(246), C(257), H(242), and C(238) atoms. In this table, can be observed that these sites have an excess of positive atomic charge, or a deficit of electrons as expected. Another result obtained is that the hydrogen and carbon atoms within the LUMO are the most likely to receive electrons from external molecules.
On the other hand, according to the largest Fukui indices, it is found that sites most likely to suffer reactions electrophilic are the sites occupied by 0(528), C(533), C(534), and 0(542) atoms. It is observed that in these atoms the largest amount of negative atomic charge is concentrated, that is to say, it has an excess of electrons, and it is from these sites that electrons are transferred to an external electrophilic molecule. In addition, we can see that the atoms within the HOMO orbital most likely to carry out electrophilic reactions with external molecules are those of oxygen and carbon.
Our results indicate that the largest Fukui indices along the RBD molecule for radical attacks are located outside the HOMO and LUMO frontier orbitals, and they correspond to atoms O(112), S(417), O(684), and O(729). It is noteworthy that the Mulliken atomic charge that is concentrated in these sites is negative and that it is the oxygen atoms outside the frontier orbitals that are the most likely to suffer this type of attack.
Through the DFT calculation of the Mulliken population analysis [17], we found that the amino acids with the highest amount of positive or negative charge are 477S, 478T, 484E, 501N with Mulliken atomic charges of -0.032 au, 0.025 au, -0.213 au, -0.102 au, respectively. These results indicate that ionic or Van der Waals interactions with external molecules can be carried out at the sites of these amino acids.
On the other hand, Graphs 1,2 present the values of the Fukui indices of the 768 atoms, belonging to the 50 amino acids that make up the RBD, for nucleophilic and electrophilic attacks, respectively.
In Graph 1, it can be seen that the amino acids that have sites with the highest Fukui index of nucleophilic attack are L455; F456; D467, which is inside the LUMO orbital; I468, located within the LUMO orbital; F486; Y489, within the HOMO orbital; Q493; S494; N501; G502; V503; Y505.

Graph 1: Value of the Fukui index for nucleophilic attack of the 786 atoms of the 50 amino acids that make up the RBD.
In Graph 2, it is found that the amino acids that have an atom with the highest Fukui index for electrophilic attack are: L455; F456; R466; D467, within the LUMO orbital; I468, within the LUMO orbital; C480; F486; Y489, within the HOMO orbital; Q493; S494; P499; N501; G502; V503; Y505.
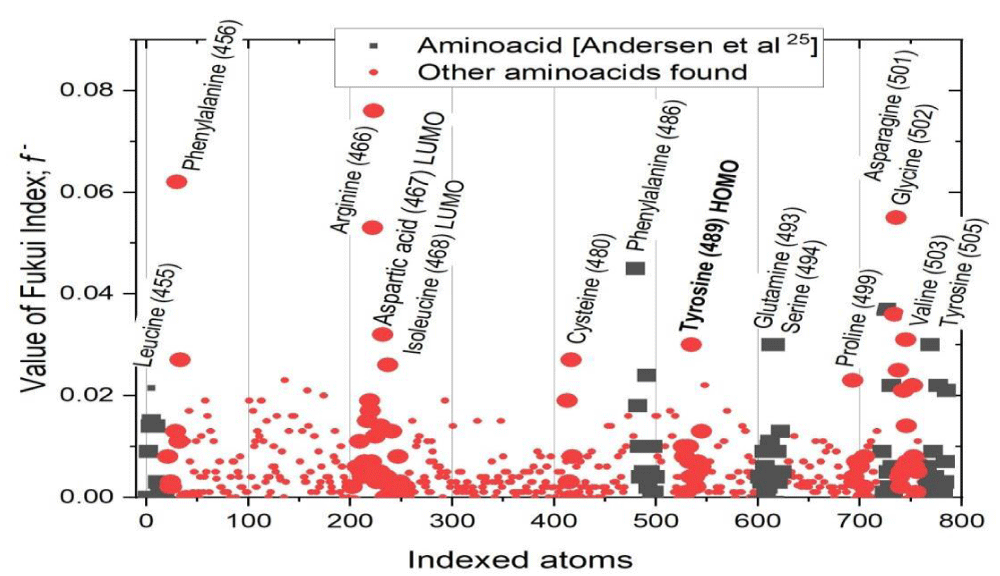
Graph 2: Value of the Fukui index for electrophilic attack of the 786 atoms of the 50 amino acids that make up the RBD.
These 15 amino acids have the highest probability and affinity to bind with the ACE2 of the human organism, and cause contagion. Of these, amino acids L455, F486, Q493, S494, N501, and Y505 have been reported since 2020 as the six RBD amino acids to bind to ACE2 [27]. Thus, in this study, another nine amino acids have been found that can cause the binding between RBD and ACE2, which represents a new contribution to the knowledge of the RBD of wild-type SARS-CoV-2.
Finally, Figure 4 shows the IR absorption spectrum of the RBD structure. It can be seen that the spectrum shows the elongations corresponding to the amino groups at around 3456 cm-1. At 2700 to 3000 cm-1, the elongations corresponded to the CH, CH2, and CH3 groups, while at around 1100 to 1200 cm-1 the bands corresponding to the CO were observed.
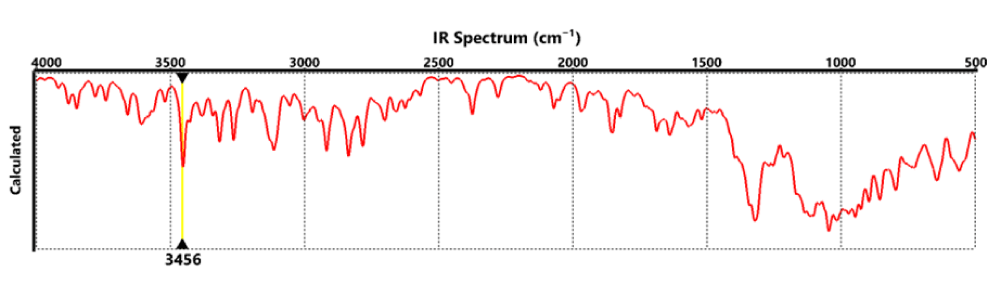
Figure 4: IR absorption spectrum of the RBD.
Current drugs with the purpose facing respiratory diseases mainly caused by SARS-CoV-2 are harmful to senior citizens or human beings with a sensible immunological system [28,29] These motivate the research of new active antiviral compounds, being the main strategy of the molecular modeling method [30,31]. Showing the first step for a magnificent research of the best inhibitors, briefly, this work gives unchangeable data for the due of the molecular docking study and generates the first generations of the possible spike selective inhibitors.
Using computational modeling and simulation techniques based on density functional theory, the most stable structure of a molecular model of the receptor binding domain (RBD) of the spike protein of the wild-type or original-type SARS-CoV-2 virus was obtained. Our results indicate that the LUMO molecular orbital is in the region occupied by the amino acids 467 Aspartic, 468 Isoleucine, 469 Serine, and 470 Threonine, with its calculated eigenvalue being -2.167 eV. While the HOMO molecular orbital is located in the region of amino acid 489 Tyrosine with an energy of -5,559 eV.
The results of the HOMO and LUMO orbitals, together with the values of the Fukui indices and electric charge, lead us to affirm that the electrophilic attacks with the greatest probability of occurring will be carried out in the 0(528), C(533), C(534) and 0(542) atoms, while the nucleophilic attacks will occur with highest probability in H(246), C(257), H(242) and C(238) atoms. Radical attacks are most likely to occur on 0(112), S(417), O(684) and O(729) atoms.
The DFT results of the global reactivity indices (chemical potential, chemical hardness, and global electrophilicity) indicate that the RBD of the SARS-CoV-2 protein S is more prone to receiving electrons from external molecules with a chemical potential greater than 0.255 eV, and to transfer electrons to external molecules with chemical potential less than 0.255 eV, this result can be related to the high degree of contagion of the SARS-CoV-2 virus; likewise, chemical hardness indicates that the RBD globally maintains its atomic and molecular structure during chemical reactions or polar or electrostatic interactions with other systems; On the other hand, the electrophilicity value indicates that the energy power of the RBD studied remains almost constant after chemical reactions with other external molecules, which justifies the high virulence of SARS-CoV-2 in the human body.
The Mulliken charge distribution indicates that the highest concentrations of positive and negative charge are found in the zones 477S, 478T, 484E, 501N with Mulliken atomic charges of -0.032 au, 0.025 au, -0.213 au, -0.102 au, respectively, which are the most likely to carry out polar, electrostatic or Van der Walls interactions with external systems.
Finally, the IR absorption spectrum of the RBD structure indicates that the spectrum shows the elongations corresponding to the amino groups at around 3456 cm-1. At 2700 to 3000 cm-1 the elongations corresponded to the CH, CH2, and CH3 groups, while at around 1100 to 1200 cm-1 the bands correspond to the CO group.
In this study, another nine amino acids have been found that can cause the binding between RBD and ACE2, which represents a new contribution to the knowledge of the RBD of wild-type SARS-CoV-2.
Authors’ contributions
Ernesto López-Chávez and Alberto García-Quiroz wrote the original manuscript.
All authors prepared figures and tables.
All authors performed and analyzed the calculations.
All authors reviewed the manuscript.
Ernesto López-Chávez led the entire team.
Funding
Dr. Alberto García Quiroz greatly thanks the UACM for all the infrastructure and economic support, among other support, for the resources of the CCyT-2021-3 internal project of the College of Science and Technology. All authors also appreciate the financial support provided by the National Polytechnic Institute, through the EDD and COFAA scholarships, and CONAHCyT through the economic stimulus of the National System of Researchers.
Availability of data and materials
The data sets used, and the parameters used in the launch of the calculations can be accessed by consulting the Theoretical Methodology section of the manuscript and the Supplementary information in the annexes.
- Yan ZP, Yang M, Lai CL. COVID-19 Vaccines: A Review of the Safety and Efficacy of Current Clinical Trials. Pharmaceuticals (Basel). 2021 Apr 25;14(5):406. doi: 10.3390/ph14050406. PMID: 33923054; PMCID: PMC8144958.
- Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021 Mar;19(3):141-154. doi: 10.1038/s41579-020-00459-7. Epub 2020 Oct 6. Erratum in: Nat Rev Microbiol. 2022 May;20(5):315. PMID: 33024307; PMCID: PMC7537588.
- V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021 Mar;19(3):155-170. doi: 10.1038/s41579-020-00468-6. Epub 2020 Oct 28. PMID: 33116300; PMCID: PMC7592455.
- Khare P, Sahu U, Pandey SC, Samant M. Current approaches for target-specific drug discovery using natural compounds against SARS-CoV-2 infection. Virus Res. 2020 Dec;290:198169. doi: 10.1016/j.virusres.2020.198169. Epub 2020 Sep 24. PMID: 32979476; PMCID: PMC7513916.
- Jawad B, Adhikari P, Podgornik R, Ching WY. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J Chem Inf Model. 2021 Sep 27;61(9):4425-4441. doi: 10.1021/acs.jcim.1c00560. Epub 2021 Aug 24. PMID: 34428371.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020 Apr 16;181(2):281-292.e6. doi: 10.1016/j.cell.2020.02.058. Epub 2020 Mar 9. Erratum in: Cell. 2020 Dec 10;183(6):1735. PMID: 32155444; PMCID: PMC7102599.
- Kang YF, Sun C, Zhuang Z, Yuan RY, Zheng Q, Li JP, Zhou PP, Chen XC, Liu Z, Zhang X, Yu XH, Kong XW, Zhu QY, Zhong Q, Xu M, Zhong NS, Zeng YX, Feng GK, Ke C, Zhao JC, Zeng MS. Rapid Development of SARS-CoV-2 Spike Protein Receptor-Binding Domain Self-Assembled Nanoparticle Vaccine Candidates. ACS Nano. 2021 Feb 23;15(2):2738-2752. doi: 10.1021/acsnano.0c08379. Epub 2021 Jan 19. PMID: 33464829.
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020 May;581(7807):215-220. doi: 10.1038/s41586-020-2180-5. Epub 2020 Mar 30. PMID: 32225176.
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI; Sheffield COVID-19 Genomics Group; McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020 Aug 20;182(4):812-827.e19. doi: 10.1016/j.cell.2020.06.043. Epub 2020 Jul 3. PMID: 32697968; PMCID: PMC7332439.
- Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2022 Jan;23(1):3-20. doi: 10.1038/s41580-021-00418-x. Epub 2021 Oct 5. PMID: 34611326; PMCID: PMC8491763.
- Yamasoba D, Kimura I, Nasser H, Morioka Y, Nao N, Ito J, Uriu K, Tsuda M, Zahradnik J, Shirakawa K, Suzuki R, Kishimoto M, Kosugi Y, Kobiyama K, Hara T, Toyoda M, Tanaka YL, Butlertanaka EP, Shimizu R, Ito H, Wang L, Oda Y, Orba Y, Sasaki M, Nagata K, Yoshimatsu K, Asakura H, Nagashima M, Sadamasu K, Yoshimura K, Kuramochi J, Seki M, Fujiki R, Kaneda A, Shimada T, Nakada TA, Sakao S, Suzuki T, Ueno T, Takaori-Kondo A, Ishii KJ, Schreiber G; Genotype to Phenotype Japan (G2P-Japan) Consortium; Sawa H, Saito A, Irie T, Tanaka S, Matsuno K, Fukuhara T, Ikeda T, Sato K. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022 Jun 9;185(12):2103-2115.e19. doi: 10.1016/j.cell.2022.04.035. Epub 2022 May 2. PMID: 35568035; PMCID: PMC9057982.
- Qiao B, Olvera de la Cruz M. Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites. ACS Nano. 2020 Aug 25;14(8):10616-10623. doi: 10.1021/acsnano.0c04798. Epub 2020 Aug 4. PMID: 32806067.
- Gao K, Wang R, Chen J, Cheng L, Frishcosy J, Huzumi Y, Qiu Y, Schluckbier T, Wei X, Wei GW. Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2. Chem Rev. 2022 Jul 13;122(13):11287-11368. doi: 10.1021/acs.chemrev.1c00965. Epub 2022 May 20. PMID: 35594413; PMCID: PMC9159519.
- Wang Q, Wang L, Zhang Y, Zhang X, Zhang L, Shang W, Bai F. Probing the Allosteric Inhibition Mechanism of a Spike Protein Using Molecular Dynamics Simulations and Active Compound Identifications. J Med Chem. 2022 Feb 24;65(4):2827-2835. doi: 10.1021/acs.jmedchem.1c00320. Epub 2021 Aug 20. PMID: 34415156.
- Padhi AK, Rath SL, Tripathi T. Accelerating COVID-19 Research Using Molecular Dynamics Simulation. J Phys Chem B. 2021 Aug 19;125(32):9078-9091. doi: 10.1021/acs.jpcb.1c04556. Epub 2021 Jul 28. PMID: 34319118.
- Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020 Apr;26(4):450-452. doi: 10.1038/s41591-020-0820-9. PMID: 32284615; PMCID: PMC7095063.
- Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can J Phys. 1980; 58:1200-1211. http://dx.doi.org/10.1139/p80-159
- Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B Condens Matter. 1992 Jun 15;45(23):13244-13249. doi: 10.1103/physrevb.45.13244. PMID: 10001404.
- Becke AD. A multicenter numerical integration scheme for polyatomic molecules. J Chem Phys. 1988; 88:2547-2553 https://doi.org/10.1063/1.454033
- Delley B. Theoretical and Computational Chemistry, 2, 221-254, Volume book: Modern density functional theory: a tool for chemistry, edited by J.M. Seminario, P. Politzer, Elsevier Science: Amsterdam, ISBN 0-444-82171-6. https://doi.org/10.1016/S1380-7323(05)80037-8
- Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys Rev Lett. 1996; 77: 3865. Erratum Phys. Rev. Lett. 78: 1396. https://doi.org/10.1103/PhysRevLett.78.1396
- Qi L, Fa-tang L. Recent advances in molecular oxygen activation via photocatalysis and its application in oxidation reactions. Chem Engi J. 2021; 421: 129915. https://doi.org/10.1016/j.cej.2021.129915
- Noorizadeh S, Taghipour KA. A regioselectivity descriptor based on atomic Weizsäcker kinetic energy. Chem Phys Lett. 2021; 770:138455. https://doi.org/10.1016/j.cplett.2021.138455
- Parr RG, Yang W. Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc. 1984; 106(14): 4049–4050 https://doi.org/10.1021/ja00326a036
- Parr RG, Yang W. Density Functional Theory of Atoms and Molecules. Oxford University Press, N.Y. USA, 1989; https://doi.org/10.1002/qua.560470107
- Parr RG, Donnelly RA, Levy M, Palke WE. Electronegativity: the density functional viewpoint. J Chem Phys. 1978; 68:3801–3807 http://dx.doi.org/10.1063/1.436185
- Mulliken RS. Electronic population analysis on LCAO-MO molecular wave functions. I and Electronic population analysis on LCAO-MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J Chem Phys. 1955; 10(23): 1833-1846. https://doi.org/10.1063/1.1740588
- Boretti A. Favipiravir use for SARS CoV-2 infection. Pharmacol Rep. 2020; 72(6): 1542-1552. https://doi.org/10.1007/s43440-020-00175-2.
- Valle C, Martin B, Touret F, Shannon A, Canard B, Guillemot JC, Coutard B, Decroly E. Drugs against SARS-CoV-2: What do we know about their mode of action? Rev Med Virol. 2020 Nov;30(6):1-10. doi: 10.1002/rmv.2143. Epub 2020 Aug 11. PMID: 32779326; PMCID: PMC7435512.
- Xia S, Chen E, Zhang Y. Integrated Molecular Modeling and Machine Learning for Drug Design. J Chem Theory Comput. 2023 Nov 14;19(21):7478-7495. doi: 10.1021/acs.jctc.3c00814. Epub 2023 Oct 26. PMID: 37883810; PMCID: PMC10653122.
- Gupta R, Srivastava D, Sahu M, Tiwari S, Ambasta RK, Kumar P. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol Divers. 2021 Aug;25(3):1315-1360. doi: 10.1007/s11030-021-10217-3. Epub 2021 Apr 12. PMID: 33844136; PMCID: PMC8040371.